Thalassemia is a group of genetic blood disorders where the synthesis of one of the polypeptide chains that form hemoglobin is decreased, resulting in abnormal formation of hemoglobin and destruction of red blood cells, leading to anemia. There are 2 major types: alpha thalassemia occurs when genes related to alpha globin protein are missing or mutated, and beta thalassemia occurs when genetic defects affect beta globin protein production.
Common clinical features of alpha thalassemia include anemia with skin pallor, weakness, and fatigue.
The most severe form of alpha thalassemia is hemoglobin Bart hydrops fetalis (Hb Bart) syndrome. Hb Bart syndrome is characterized by hydrops fetalis, a condition where excess fluid builds in the body prior to birth, resulting in hepatosplenomegaly, cardiac abnormalities, and genitourinary defects. Most neonates with this form of thalassemia are stillborn or die shortly after birth. Complications in the birthing parent include preeclampsia, premature delivery, and abnormal bleeding.
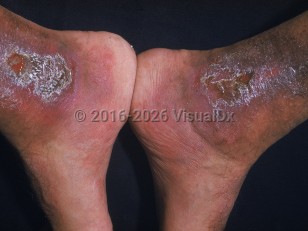
Hemoglobin H (HbH) disease is another form of alpha thalassemia that causes mild-to-moderate anemia, hepatosplenomegaly, and jaundice. In some cases, patients may experience bone changes due to extra-medullary hematopoiesis such as jaw overgrowth or prominent forehead. Onset is typically in early childhood, and patients usually live into adulthood.
Alpha thalassemia occurs more frequently in residents of Southeast Asia, North Africa, India, Central Asia, the Middle East, and Mediterranean countries. Caused by HBA1 and HBA2 gene deletions, it has a complex inheritance pattern, and offspring may inherit if both parents are missing at least one alpha globin allele.
Treatment is dependent on the severity of disease.
Alpha thalassemia
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
D56.0 – Alpha thalassemia
SNOMEDCT:
68913001 – Alpha thalassemia
D56.0 – Alpha thalassemia
SNOMEDCT:
68913001 – Alpha thalassemia
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:02/20/2018
Last Updated:05/12/2025
Last Updated:05/12/2025
Alpha thalassemia