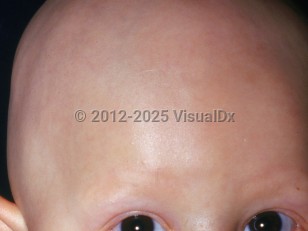
Atrichia with papular lesions (APL) is a rare autosomal recessive syndrome associated with irreversible hair loss and widespread papular lesions. A mutation in the zinc finger domain of the human hairless HR gene on chromosome region 8p12 produces a dysplasia of the pilosebaceous system.
In APL, the proximal and outer root sheath of hair follicles undergoes apoptosis and disintegration at the end of the anagen phase. Patients are born with scalp and body hair but develop irreversible hair loss beginning at a few months of age, with a frontal to posterior progression on the scalp. In some cases, patients are born hairless. Papular lesions erupt within the first year of life, most prominently under the midline of the eye, face, and extremities. Papular lesions are made of keratin cysts associated with malformation of the pilosebaceous system.
Atrichia congenita - Hair and Scalp
See also in: OverviewAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q84.0 – Congenital alopecia
SNOMEDCT:
403798006 – Atrichia congenita
Q84.0 – Congenital alopecia
SNOMEDCT:
403798006 – Atrichia congenita
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/11/2022
Atrichia congenita - Hair and Scalp
See also in: Overview