Behçet syndrome is relatively uncommon and is more prevalent in Turkey, Korea, Japan, the Middle East, and Mediterranean countries. While there is an equal sex distribution overall, there is male predominance in Middle Eastern and Mediterranean countries and female predominance in Japan and Korea. Severe disease is more commonly seen in males. The onset of symptoms typically occurs during the third or fourth decades of life. Rarely, it can present during childhood, and usually in children with a positive family history of the syndrome.
Behçet syndrome is strongly associated with the HLA-B51 allele, which is present in more than 80% of Asian patients with Behçet syndrome. The pathogenesis of Behçet syndrome involves circulating immune complexes and neutrophils causing vascular injury. Recently, single nucleotide polymorphisms in interleukin (IL)-10 and IL-23R were found to be associated with developing the disease. Triggers in genetically predisposed patients include infectious agents, histamine-releasing foods, poor oral hygiene, and stress.
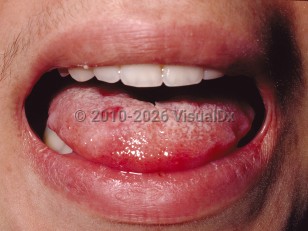
Clinically, Behçet syndrome is characterized by recurrent oropharyngeal and genital ulcers, and uveitis. Recurrent aphthous stomatitis is the initial presenting sign in up to 80% of patients and can lead to dysphagia and odynophagia. Genital ulcers tend to recur less frequently but are more prone to scar formation. Potential complications include fistulae in females and epididymo-orchitis in males.
Ocular disease is characterized by episodes of bilateral, nongranulomatous anterior and/or posterior uveitis; it is seen more often in males. It usually appears 2-3 years after the onset of oral and/or genital ulcers, although it can be the initial manifestation in up to 20% of cases. Approximately 1 in 4 patients with ocular disease develop blindness.
Cutaneous manifestations include erythema nodosum-like lesions, pustules, erythema multiforme-like lesions, Sweet syndrome-like lesions, subcutaneous thrombophlebitis, and palpable purpura.
Other systemic manifestations include:
- Musculoskeletal – Arthralgia; nonerosive mono- or polyarthritis is seen in 45%-60% of patients, affecting the knees, ankles, hips, elbows, and wrists.
- Neurologic – Occurs in approximately 10% of patients, often multiple years after the onset of other symptoms. May include cerebellar dysfunction, cranial nerve palsies, cerebral venous thrombosis, arterial vasculitis, aseptic meningitis, and encephalitis.
- Vascular – Deep vein thromboses and pulmonary artery aneurysms are seen.
- Cardiac – Including pericarditis, myocarditis, mitral valve prolapse, intracardiac thrombosis, endomyocardial fibrosis, cardiomyopathy, and coronary artery lesions.
- Gastrointestinal – Mucosal inflammation and ulceration (particularly ileocecal region); inflammatory bowel disease-like symptoms (eg, diarrhea, abdominal pain, gastrointestinal bleeding).