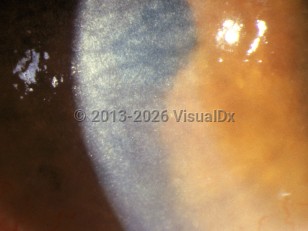
Cystinosis is a clinically heterogeneous disorder with widespread organ damage resulting from tissue accumulation of cystine crystals. The most serious damage occurs in the kidney and may result in end-stage disease. However, other organs such as the thyroid and pancreas are often damaged as well.
Mutations in the CTNS gene are responsible for all cases of cystinosis, even though there can be considerable heterogeneity in clinical presentation. Individuals are generally grouped into 1 of 3 categories of disease. The most common and severe form is known as infantile or nephropathic cystinosis. These patients may have Fanconi-type renal tubular disease at an age as early as 6 months, and some patients require a renal transplant in the first decade of life. Poor feeding with failure to thrive are also evident in the first year of life. Retarded physical growth in the range of 50% of normal is evident in the same period. Hypophosphatemic rickets may be a consequence of untreated renal disease. Cystine crystals may be seen in the cornea at this time as well, and their presence causes significant sensitivity to light (photophobia). The retinal photoreceptors are damaged early, and patients may experience significant loss of vision as youngsters. Retinal pigment clumping and areas of hypopigmentation occur in the fundus periphery and progress centrally.
Intermediate or juvenile cystinosis is far less common, and the onset of clinical manifestations is often delayed until the second decade of life. Nephropathy is usually present but may be so mild that it can remain undetected until late stages. Electrolyte imbalance, growth retardation, and ocular symptoms may also escape detection for a decade or more. However, renal failure always occurs, usually by the third decade of life. These patients also have corneal crystals and photophobia and sometimes experience painful recurrent corneal erosions. Photoreceptor damage leading to vision loss is common. The pigmentary changes in the retina are similar to those in the infantile form of the disease.
Some individuals do not develop renal damage and have photophobia as their only symptom. This form of cystinosis is sometimes called ocular, non-nephropathic, or adult disease. Patients with this form of disease do not suffer photoreceptor damage or have retinal pigmentary changes. Corneal crystals cause photophobia, which may be the only symptom. Recurrent corneal erosions often occur.
Individuals with early-onset disease who survive into the third decade of life or later may develop late abnormalities such as myopathy, gastrointestinal dysfunction, cardiovascular disease, and central nervous system damage.
Cystinosis - External and Internal Eye
See also in: OverviewAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E72.04 – Cystinosis
SNOMEDCT:
190681003 – Cystinosis
E72.04 – Cystinosis
SNOMEDCT:
190681003 – Cystinosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/12/2022
Cystinosis - External and Internal Eye
See also in: Overview