EGPA has an estimated incidence of 0.1-3 cases per million and a prevalence of 11-18 cases per million worldwide. It does not have a clear sex predominance. The mean age at diagnosis is 48 years. Disease onset in childhood or adolescence is possible but extremely rare. Those affected in younger age groups tend to have a more aggressive disease course with prominent pulmonary and cardiovascular complications.
The clinical course of EGPA typically develops in 3 sequential but overlapping phases: a prodromal atopic phase, an eosinophilic phase, and finally a vasculitic phase.
- The prodromal phase frequently occurs in the second to third decade of life and may last months to years, with asthma as the main manifestation (96%-100% of patients). The asthma of EGPA is typically difficult to control with conventional treatments. Also common in this phase are allergic rhinosinusitis and nasal polyposis.
- The eosinophilic phase is marked by an elevated eosinophil count and eosinophil infiltration into multiple organs including the heart, lungs, and gastrointestinal tract.
- The vasculitic phase, typically occurring a decade or more after the onset of the prodromal phase, is characterized by a small-to-medium vessel necrotizing vasculitis with granuloma formation.
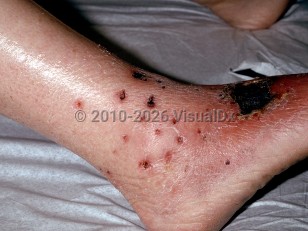
Cutaneous findings are common, especially during the vasculitic phase of EGPA, and can be polymorphic, including erythema, urticaria, purpura, tender nodules, and necrosis. Cranial symptoms include otitis media, sensorineural hearing loss, anterior scleritis, episcleritis, and facial-nerve palsy.
The exact mechanism of pathogenesis for EGPA is currently unknown. However, it is generally thought to be due to a dysregulation of immune function. Research indicates that eosinophil infiltration and antineutrophil cytoplasmic antibody (ANCA) (proteinase 3 antibody)-induced endothelial damage may be involved in the underlying disease mechanism. About half of patients with EGPA have positive ANCA. Recently, it has been suggested that 2 distinct phenotypes of EGPA are present and depend on the presence or absence of ANCA. Several medications (eg, leukotriene modifying agents, omalizumab) have also been found to be associated with the apparent onset of EGPA; however, causal relationships have not been established, and it is likely that these medications only served to unmask the underlying disease.