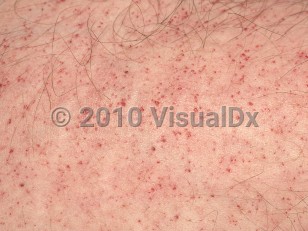
While any organ may be affected by this disorder, the kidneys, skin, heart, and brain are most commonly affected. Deposits in the vascular endothelium lead to vascular occlusion and thrombosis. Renal deposits lead to proteinuria and renal failure. Dysfunctional valves and cardiomyopathy result from cardiac deposits. Characteristic skin lesions are angiokeratomas, which are dark red to purple macules or papules located in clusters in the swimsuit area (between the umbilicus and the knees) or more diffusely. Angiokeratomas usually develop initially in childhood and tend to increase in number with time as undegraded lipids continue to accumulate in affected organs.
Signs and symptoms are myriad and highly variable. Pain is one of the earliest symptoms, and the most incapacitating, starting with acroparesthesia and a burning pain in the hands and feet, often in the first decade of life. A more severe form of pain, known as Fabry crisis, begins in the hands and feet and radiates proximally into the limbs. This pain may be initiated or exacerbated by factors such as heat, cold, fatigue, stress, illness, or exercise. Episodes of pain may last from a few minutes to hours or even days. Episodes of Fabry crisis tend to decrease with advancing age. Chronic pain may have an even greater impact on the patient's psychosocial wellbeing.
The second most common group of symptoms includes abdominal pain, nausea, vomiting, constipation, and diarrhea starting in early childhood. Additional symptoms include joint pain, intolerance to heat and exercise due to hypohidrosis or anhidrosis, dry eyes, and dry mouth. Hearing loss and tinnitus, aphasia, dysarthria, diplopia, vertigo, seizures, and valve disease occur in adulthood. The disease most commonly culminates in progressive cardiomyopathy and left heart failure, impaired renal function, and stroke, usually when patients are in the third to fifth decade of life. Given the phenotypic variability of this condition, specific disease manifestations may present earlier in life due to severe disease and/or very low enzymatic activity.
As Fabry disease is an inherited disease, the onset of symptoms begins in early childhood. However, given the nonspecific and variable presentation of symptoms, a timely diagnosis is difficult and affected individuals are commonly misdiagnosed for many years. Despite a median age of onset of symptoms within the first decade of life for males, the median age of diagnosis is in the second to third decade of life. The median age of diagnosis in females can be more than 5 years later than affected males, although the median age of onset in females is also typically later in life. Once an individual is diagnosed with Fabry disease, potentially affected family members are often diagnosed earlier in life while still asymptomatic due to more vigilant screening or testing.
Fabry disease, if untreated, generally progresses as outlined below. However, due to the heterogeneous nature of this disease, it is important to remember that the clinical signs and symptoms may vary among patients.
- Early childhood sees the onset of burning pain of the hands and feet between the ages of 5 and 7 years. Abdominal pain and diarrhea, hypohidrosis, poor growth, and then angiokeratomas develop between the ages of 5 and 13 years.
- The second cluster of symptoms begins in early adulthood, with impairment of renal function beginning as proteinuria.
- Lastly comes the onset of cerebrovascular manifestations, including conduction disturbances and progressively worsening renal function.
- Chronic renal failure and cardiac disease are the most frequent causes of death in the third to fifth decades of life. Cardiac symptoms may include chest pain, shortness of breath, syncope, heart palpitations, or reduced exercise tolerance.
The disease affects from 1:40 000 to 1:117 000 individuals, primarily hemizygous males. Heterozygous females may experience variable symptoms ranging from severe disease to absent or minimal clinical manifestations. Average lifespan of affected males is usually about 50-60 years, whereas the average life expectancy for females is just after age 75.
Classic (or type I) Fabry disease is most commonly characterized by alpha-Gal A enzyme activity that is < 1% of the normal mean and occurs predominantly in males. The onset of symptoms is almost always present in childhood or early adolescence in individuals with the classic variant, and symptoms tend to be more severe. Non-classic, or late-onset, Fabry disease is characterized by affected individuals with alpha-Gal A enzyme activity that ranges between 5% and 30% of the normal mean. Individuals with late-onset disease may not experience significant accumulation of toxic lipids in all of the organs commonly affected in patients with classic disease. Rather, late-onset Fabry disease may be associated with disease manifestations limited to fewer organ systems in late adulthood.