The familial partial lipodystrophies (FPLD) are a heterogenous group of congenital lipodystrophy disorders with several distinct phenotypes. FPLD has an estimated approximate worldwide prevalence of 1.67-2.84 cases per million population. Lipodystrophies are rare and occur in approximately 1.3-4.7 cases per million. There are 6 distinctive different types of FPLD (FPLD1 to FPLD6), as well as other partial lipodystrophies associated with rare genetic syndromes.
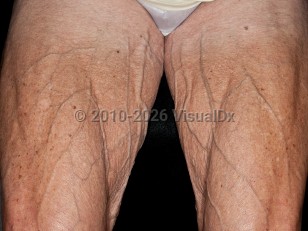
The most prevalent FPLD, with over 500 patients reported, is Dunnigan syndrome (FPLD2). It is characterized by a normal distribution of body fat in childhood with a gradual, progressive lipoatrophy of the upper and lower extremities beginning at puberty, resulting in a muscular appearance. Some patients may gain excess fat in the face and chin as well as the neck, leading to a cushingoid appearance. Less commonly, excess fat may also accumulate on the trunk and in the vulvar area.
Metabolic complications may occur later in life, including diabetes mellitus, hypertriglyceridemia, low levels of high-density lipoprotein cholesterol (HDL), and atherosclerotic disease. These are more common in women with Dunnigan syndrome than in affected men. Fatty liver and acute pancreatitis caused by marked hypertriglyceridemia may also occur. Acanthosis nigricans and polycystic ovarian disease are uncommon associated findings.
Dunnigan-type familial partial lipoatrophy results from an autosomal dominant missense mutation in the LMNA gene, which encodes 2 major proteins: lamin A and lamin C. Lamins are intermediate filaments that interact with nuclear envelope proteins, and it is believed that mutations that affect their function lead to lipoatrophy caused by premature adipocyte apoptosis.
Another less common FPLD phenotype is due to an autosomal dominantly inherited mutation in the peroxisome proliferator-activated receptor-gamma (PPARG) gene (FPLD3). This gene encodes the protein PPARG, which is a "master regulator" of adipogenesis and enhances lipogenesis without creating larger adipocytes. It also promotes subcutaneous visceral fat differentiation. This mutation, reported in about 20 families, is associated with a milder phenotype, with the onset of lipodystrophy beginning in the second decade or later in life. Unlike patients with Dunnigan-type lipodystrophy, excess fat deposition in areas such as the face, neck, trunk, and vulva does not occur. In general, patients with PPARG mutations develop more severe metabolic derangements compared to those with LMNA mutations.
Kobberling syndrome (FPLD3) patients clinically have loss of adipose tissue in their buttocks and limbs beginning in childhood / adolescence, with following subcutaneous and visceral abdominal fat accumulation on the trunk. Fat accumulation may also occur in the face or neck. Patients with Kobberling syndrome are likely to have increased blood pressure, lipid parameters, and insulin resistance (ie, elevated blood sugar levels). Acanthosis nigricans, acrochordons, and hirsutism may be significantly more present in patients with Kobberling syndrome than Dunnigan syndrome. Kobberling syndrome may be due to multiple pathogenetic mutations other than PPARG or LMNA.
Other notable gene mutations of phenotypic FDLP variants include PLIN1 (perilipin 1)-related FPLD or FPLD4, CIDEC (cell death inducing DFFA like effector C)-related FPLD or FPLD5, LIPE (hormone sensitive lipase)-related FPLD or FPLD6, and CAV1 (caveolin 1)-related FPLD, and AKT2 (protein kinase B)-related FPLD.
Mandibuloacral dysplasia (MAD) is another extremely rare type of FPLP and is inherited in an autosomal recessive fashion. It is characterized by lipodystrophy associated with skeletal abnormalities including hypoplasia of the mandible and clavicle.
Familial partial lipodystrophy
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E88.1 – Lipodystrophy, not elsewhere classified
SNOMEDCT:
49292002 – Familial partial lipodystrophy
E88.1 – Lipodystrophy, not elsewhere classified
SNOMEDCT:
49292002 – Familial partial lipodystrophy
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:01/03/2022
Last Updated:01/04/2022
Last Updated:01/04/2022
Familial partial lipodystrophy