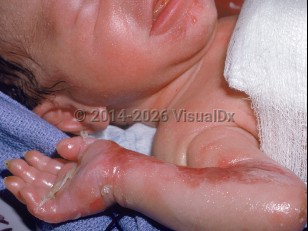
Epidermolysis bullosa simplex (EBS) is a heterogeneous group of inherited blistering disorders with 4 common subtypes. Of these, generalized severe EBS (EBS-gen/sev; formerly known as Dowling-Meara EBS [EBS-DM] and simplex herpetiformis) is one of the more severe generalized forms. EBS-gen/sev results from an autosomal dominant defect in the genes encoding either keratin-5 or -14, constituents of intermediate filaments that anchor the keratinocyte cytoskeleton to the desmosomal plaque. Disruption within cells of the basal layer of the epidermis occurs, leading to blister formation from even the slightest trauma.
EBS-gen/sev accounts for approximately 25% of EBS cases. In EBS-gen/sev, generalized blistering occurs, often with blisters in a grouped, herpetiform, arcuate, or serpiginous pattern. Onset is typically at birth. The oral mucosa is commonly involved, with blistering of the oral and esophageal mucosa during early infancy or the neonatal period, making feeding difficult. Localized atrophic scarring, milia, and nail dystrophy are commonly seen, especially as the child gets older, that may resemble dystrophic EB. In late childhood, repeated blistering of the palms and soles leads to the development of keratoderma, a characteristic finding. As in all other types and subtypes of inherited EB, blistering worsens during warmer weather. Curiously, EBS-gen/sev patients may have temporary improvement in skin disease activity during periods of high fever. Laryngeal involvement can cause stenosis, strictures, and hoarseness. Some patients with EBS-gen/sev may experience improvement in blister activity during mid-adulthood, and rarely some of these keratodermas may partially regress.
Extracutaneous complications often include blistering of the oral cavity. Progressive, potentially life-threatening occlusion of the upper airway as a result of repeated blistering at or above the level of the vocal cords has been reported. Contractures of the arms and legs may be seen, as well as growth retardation and anemia, although the latter are uncommon. Most patients with EBS-gen/sev live a normal lifespan, with a minority of the most severely affected dying during early infancy from overwhelming sepsis or failure to thrive. Squamous cell carcinoma appears to be less of a concern in patients with EBS than in other forms of EB.
Related topics: EB acquisita, junctional EB, Kindler syndrome
Generalized severe epidermolysis bullosa simplex in Infant/Neonate
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q81.0 – Epidermolysis bullosa simplex
SNOMEDCT:
90496008 – Generalized epidermolysis bullosa simplex
Q81.0 – Epidermolysis bullosa simplex
SNOMEDCT:
90496008 – Generalized epidermolysis bullosa simplex
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:04/16/2024
Last Updated:05/15/2024
Last Updated:05/15/2024
Generalized severe epidermolysis bullosa simplex in Infant/Neonate