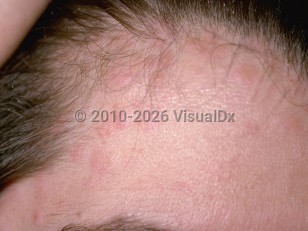
Hereditary angioedema is a rare genetic condition characterized by sudden attacks of angioedema with associated complications. Angioedema is characterized by swelling of the deep dermis and subcutaneous / submucosal tissue. It is less well-defined than the superficial dermal swelling seen with the wheals of urticaria, and the affected areas are nonpruritic. Prodromal symptoms, including a tingling sensation, fatigue, or myalgia, may occur. Angioedema is often idiopathic but may also be a reaction to drugs or food, or it may be hereditary. This summary discusses hereditary angioedema.
Hereditary angioedema follows an autosomal dominant pattern of inheritance and occurs in approximately 1 in 50 000-150 000 individuals worldwide. The condition has been reported in all races and ethnicities and affects both sexes equally. Symptoms of hereditary angioedema typically first present in the second decade of life, although they may manifest in the first decade.
Angioedema can affect any part of the body and multiple sites at once. Areas of swelling can last for up to 48-72 hours. Commonly affected areas include the face, extremities, genitalia, trunk, upper respiratory tract, and gastrointestinal tract. Most attacks are brought on by trauma or stress. Acute attacks involving upper airway swelling can be life-threatening. The nonspecific gastrointestinal symptoms of hereditary angioedema can often be erroneously attributed to more common causes, particularly in pediatric patients, as abdominal pain is a common complaint in childhood. Severe attacks may be mistaken for acute abdomen. Because of these factors, hereditary angioedema with primarily abdominal symptoms can go undiagnosed or be misdiagnosed for years.
Hereditary angioedema is caused by a deficiency or diminished function of C1 esterase inhibitor. Inherited or spontaneous (approximately 25%) mutation in one copy of the SERPING gene for C1 esterase inhibitor results in decreased quantity of C1 esterase inhibitor (type I) in about 85% of cases, or reduced function of C1 esterase inhibitor (type II) in about 15% of cases. This allows for the unregulated activation of proteolytic pathways and the unchecked downstream production of vasoactive anaphylatoxins including bradykinin. The resulting excess bradykinin production increases vascular permeability and leads to localized tissue edema. C1 esterase inhibitor deficiency also causes activation of the C1 complement system, resulting in low serum C4, which is a diagnostic feature.
A more recently described type (type III) of hereditary angioedema has been recognized, which is associated with normal measurable quantities and function of C1 esterase inhibitor. It is seen primarily in women with a personal and family history of angioedema, but no demonstrable deficiency of C1 esterase inhibitor or other complement abnormalities. It is believed that attacks may be caused by defects in other pathways that result in bradykinin overproduction. It appears to be estrogen dependent, since affected women often reported disease onset or worsening of symptoms with the use of oral contraceptives containing estrogen.
Hereditary angioedema
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
D84.1 – Defects in the complement system
SNOMEDCT:
82966003 – Hereditary angioedema
D84.1 – Defects in the complement system
SNOMEDCT:
82966003 – Hereditary angioedema
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:03/18/2020
Last Updated:01/17/2022
Last Updated:01/17/2022
Hereditary angioedema