Hyperimmunoglobulin D with periodic fever syndrome (HIDS), also known as mevalonate kinase deficiency (MKD), is a monogenic autoinflammatory syndrome characterized by recurrent fevers and systemic symptoms. HIDS is inherited in an autosomal recessive manner and is seen equally in both males and females. It is caused by a mutation in the MVK gene, which encodes for the enzyme mevalonate kinase, a key enzyme in the cholesterol biosynthesis pathway. The majority of patients have compound heterozygous mutations. Although the exact etiology of the inflammation is not fully understood, there are elevated levels of interleukin-1 beta (IL-1β) in association with inflammation.
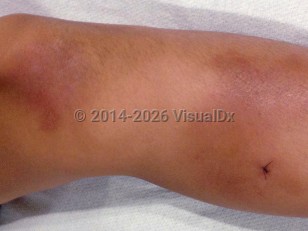
HIDS typically presents in infancy with a median age of onset of 6 months. The primary manifestation of HIDS is recurrent febrile attacks occurring at varied intervals with duration of 3-7 days. Frequent triggers of febrile attacks include vaccinations, illness, and emotional stress. The attacks may begin with a prodrome of headache and malaise followed by the onset of a high fever, often 40°C (104°F) or higher. Patients may also experience tender cervical lymphadenopathy, abdominal pain, diarrhea, vomiting, arthralgias or nonerosive arthritis, splenomegaly, oral ulcers (in up to a half of patients) or genital ulcers, and pharyngitis. Cutaneous manifestations are observed in about 70% of patients with HIDS; however, skin findings do not necessarily occur with every febrile attack. The skin findings include erythema nodosum, vasculitis, urticarial lesions, and a morbilliform eruption.
Although a few cases of associated amyloidosis have been reported, amyloidosis is not typically seen with HIDS.
Note: It is rare for symptoms to first manifest in those older than 5 years, so other periodic fever syndromes should be considered first in such cases.
Hyperimmunoglobulinemia D syndrome
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
D82.8 – Immunodeficiency associated with other specified major defects
SNOMEDCT:
403834003 – Hyper-immunoglobulin D periodic fever syndrome
D82.8 – Immunodeficiency associated with other specified major defects
SNOMEDCT:
403834003 – Hyper-immunoglobulin D periodic fever syndrome
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:04/19/2018
Last Updated:12/27/2022
Last Updated:12/27/2022
Hyperimmunoglobulinemia D syndrome