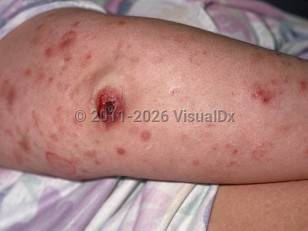
Lymphomatoid papulosis (LyP) is a recurrent, chronic, self-healing eruption that exists on a continuum with primary cutaneous anaplastic large cell lymphoma (C-ALCL). These 2 entities and their overlap cases make up the CD30-positive cutaneous lymphoproliferative disorders that represent about 25% of cutaneous T-cell lymphomas. Despite its malignant histopathologic qualities, LyP generally follows a benign clinical course. LyP and C-ALCL share many histopathologic features; morphology and disease course are important distinguishing factors.
LyP has an incidence of roughly 1.2-1.9 cases per 1 000 000 people and may occur at any age, although it is unusual in childhood. LyP is characterized by recurrent crops of pruritic, papulonecrotic, or nodular lesions that are self-healing over weeks or months. Lesions in various stages of healing are often seen. Lesions are often pruritic and may resolve with scarring. Six histopathologic subtypes are recognized by the 2018 World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) classification and are described in Best Tests. Specific subtypes of LyP do not correlate with clinical findings. Both the clinical history and histopathologic findings must be consistent with LyP for diagnosis; histopathology alone is not sufficient.
The cause of LyP is unknown, and its pathogenesis is only partially understood. Interactions between CD30 and its ligand appear to be important, and mutations in TGF-beta receptors may contribute. Identification of human T-cell lymphotropic virus type 1 (HTLV-1), herpesviruses 6, 7, and 8, and human endogenous retroviruses within LyP has been reported, but no clear relationship has been found. Several reports also suggest radiation therapy and fingolimod as triggers.
There is debate whether LyP is an inflammatory disorder or an indolent T-cell malignancy, but most authors categorize it as the latter. Most patients with LyP have chronic, self-limited eruptions. It is estimated that 15%-50% of patients with LyP will develop another lymphoproliferative disorder, such as ALCL, Hodgkin disease, or mycosis fungoides (MF). Patients with LyP should be monitored closely for these additional malignancies. A cohort study comprised of 504 LyP patients found that there was an increased risk of developing nonhematologic malignancies such as squamous cell carcinoma (SCC), melanoma, bladder cancer, and lung cancer. Further data confirming these findings are needed.
LyP has a 10-year disease-specific survival rate of close to 100%.
Lymphomatoid papulosis in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
C86.60 – Primary cutaneous CD30-positive T-cell proliferations not having achieved remission
SNOMEDCT:
31047003 – Lymphomatoid papulosis
C86.60 – Primary cutaneous CD30-positive T-cell proliferations not having achieved remission
SNOMEDCT:
31047003 – Lymphomatoid papulosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:08/07/2022
Last Updated:08/08/2022
Last Updated:08/08/2022
Lymphomatoid papulosis in Child