Hereditary PPKs are approached and classified by the pattern of hyperkeratosis: diffuse, focal (often occurring over weight-bearing areas), or punctate.
Diffuse hereditary PPK:
- Vorner (epidermolytic) PPK and Unna-Thost (nonepidermolytic) PPK are the result of keratin mutations and show waxy or verrucous, white-yellow, symmetric hyperkeratosis.
- Mal de Meleda is a rare diffuse hereditary PPK associated with SLURP1 mutations and features stocking-glove distribution of hyperkeratosis with malodor and nail changes.
- Vohwinkel syndrome (mutilating PPK) has 2 variants: the classic form associated with deafness and mutations of the connexin gene GJB2 and the loricrin variant associated with loricrin mutations and ichthyosis. The PPK shows a diffuse honeycomb pattern. Additional features include starfish-shaped keratotic plaques on dorsal hands, feet, elbows, and knees as well as constricting digital bands termed "pseudo-ainhum," which may progress to autoamputation.
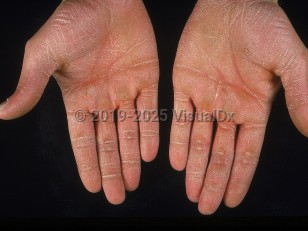
- Papillon-Lefèvre syndrome is associated with mutations in the gene that encodes cathepsin C and demonstrates diffuse PPK, periodontal disease with loss of teeth, and frequent cutaneous and systemic pyogenic infections.
- Olmsted syndrome is a hereditary disorder of mutilating PPK with periorificial plaques. Affected patients may also experience palmoplantar pruritus, diffuse alopecia, and keratosis pilaris. Onychodystrophy and digital autoamputation can be features of the disease. Autosomal dominant Olmsted syndrome has been associated with a defect of TRPV3 that may lead to erythromelalgia in some patients. Some cases of Olmsted syndrome are caused by mutations in PERP (TP53 apoptosis effector) disrupting desmosomal proteins.
- PPK has been associated with 2 cases with allelic variants in lanosterol synthase. In addition to progressive PPK, these patients had congenital alopecia and sclerodactyly (PPK-congenital alopecia syndrome type 2).
- Other diffuse hereditary PPKs include Greither syndrome, Bart-Pumphrey syndrome (PPK with knuckle pads, leukonychia, and deafness), Huriez syndrome (PPK with scleroatrophy), Clouston syndrome (hidrotic ectodermal dysplasia), diffuse nonepidermolytic PPK with sensorineural deafness, and Naxos disease (diffuse nonepidermolytic PPK with woolly hair and cardiomyopathy).
- Isolated focal PPKs (striate PPKs) are due to autosomal dominant mutations in genes encoding desmosomal proteins. Lesions favor pressure points on feet and may present as linear plaques on hands.
- Howel-Evans syndrome is associated with mutations in the TOC gene, focal weight-bearing area plantar hyperkeratosis, milder palm involvement, and development of esophageal carcinoma.
- Richner-Hanhart syndrome is associated with mutations in the gene that encodes tyrosine aminotransferase. Accumulation of tyrosine leads to focal (or diffuse) hyperkeratotic plaques on the hands, feet, elbows, and knees, corneal inflammation / ulceration, and intellectual disability in some cases. Diets low in phenylalanine and tyrosine may prevent complications.
- Focal PPK may also be seen in pachyonychia congenita type I and type II (syndromes with nail, skin, teeth, and eye anomalies), Carvajal syndrome (striate focal epidermolytic PPK with woolly hair and dilated cardiomyopathy), and skin fragility, woolly hair, and palmoplantar keratoderma associated with absence of tuftelin-1, a desmosome-associated protein.
- Punctate PPKs are characterized by autosomal dominant inheritance and multiple firm 2-8 mm papules on the palms and soles. A pattern with lesions favoring palmar creases has been identified in patients of African descent.
- Focal acral hyperkeratosis and acrokeratoelastoidosis present as 2-4 mm papules (some umbilicated) at the marginal borders of hands and feet.
- Spiny keratoderma is a rare entity characterized by multiple asymptomatic "spiny" projections of the palms and/or soles. These keratotic plugs have been likened to the spines of a music box. Both hereditary and acquired variants have been described. The hereditary form usually presents in childhood and is benign. The acquired form presents later in life and may be associated with an internal malignancy or other systemic disease.