Scleromyxedema, also known as papular mucinosis, is a primary dermal mucinosis. Although the term previously included 2 subsets of the disease (a generalized form and a localized form), scleromyxedema is now distinguished from the localized variants of lichen myxedematosus based on the different clinical and pathologic findings of these entities.
Scleromyxedema is a rare, chronic, idiopathic disorder of cutaneous mucin deposition. It affects males and females equally and presents between the ages of 30 and 80 years. More than 80% of patients have an associated paraproteinemia, typically of the immunoglobulin G (IgG)-lambda type. However, paraprotein levels do not correlate with disease activity, and thus an etiologic association remains controversial.
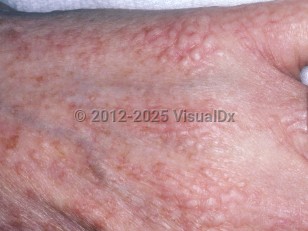
Patients present with multiple 1- to 3-mm, waxy, dome-shaped or flat-topped papules. These papules are typically widespread and symmetric and are often arranged in linear arrays. They may coalesce into plaques. Diffuse infiltration may occur, presenting with confluent lichenoid plaques, leading to a woody, sclerodermoid quality to the skin. Coalescence of infiltrative lesions on the glabella and forehead may lead to leonine facies, and infiltration on the trunk and extremities can lead to the "Shar-Pei sign" (deep furrows).
The head and neck region, upper trunk, forearms, dorsal hands, and thighs are the most commonly involved sites. Patients may experience a reduced range of motion of the hands, the upper and lower extremities, and even the mouth as a result of the disorder. Pruritus may or may not be present.
Scleromyxedema is a progressive disorder and, if untreated, it can be associated with significant morbidity and mortality. It frequently affects multiple extracutaneous organ systems, most commonly the gastrointestinal tract. Other organ systems that may be involved include the cardiovascular, pulmonary, muscular, neurologic, rheumatologic, and central nervous systems. The dermato-neuro syndrome of scleromyxedema, which begins with abrupt worsening of skin lesions, flu-like prodrome, fever, and seizures, is a potentially life-threatening encephalopathy that can lead to coma.
The diagnosis of scleromyxedema requires mucin deposition, fibroblast proliferation, fibrosis, normal thyroid function tests, and the presence of a monoclonal gammopathy. Bone-marrow evaluation in some patients may be normal, or it may range from increased numbers of plasma cells to frank multiple myeloma. Approximately 10% of patients do not have a paraproteinemia.
Related topics: cutaneous mucinosis of infancy, lichen myxedematosus
Scleromyxedema
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
L98.5 – Mucinosis of the skin
SNOMEDCT:
402468007 – Scleromyxedema
L98.5 – Mucinosis of the skin
SNOMEDCT:
402468007 – Scleromyxedema
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:02/19/2019
Last Updated:01/10/2024
Last Updated:01/10/2024
Scleromyxedema