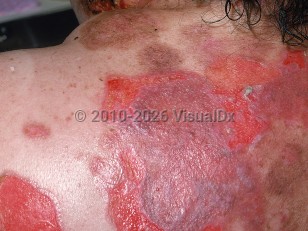
Pemphigus vulgaris (PV) is an acquired autoimmune bullous disease of the mucous membranes with or without skin involvement. It is typically characterized by the presence of circulating pathogenic autoantibodies (predominantly immunoglobulin [Ig]G4) against desmoglein, a cadherin-family, keratinocyte cell surface adhesion molecule of the desmosome, although other antibody subtypes and autoantibodies against other antigenic targets have been described. The target antigens in PV are desmoglein 3 (Dsg3) with or without desmoglein 1 (Dsg1). More than half of patients have both skin and mucosal involvement. In the mucosal-dominant type of PV, autoantibodies against Dsg3 (anti-Dsg3 Ig) are typically present, and in the mucocutaneous type, autoantibodies against Dsg1 and Dsg3 are typically present; however, variations in these patterns can be seen in the real-world setting.
The estimated incidence worldwide is 0.76-5 cases per million per year, although PV occurs in higher incidences in individuals of Jewish ancestry, as well as in certain geographic areas (Middle East, Southeastern Europe, and India). Variants of the ST18 gene have been found to confer increased risk of PV in some populations. PV is typically a disease of adults, with average onset between the ages of 40 and 60 years, but PV rarely can occur in childhood and young adulthood. There does not appear to be a consistent sex predilection.
Severe cases of PV can be life-threatening, and complications can be related to immunosuppression from drugs used to treat severe PV, secondary infections, loss of the skin barrier, and poor oral intake.
Pemphigus vulgaris in Adult
See also in: Anogenital,Oral Mucosal LesionAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
L10.0 – Pemphigus vulgaris
SNOMEDCT:
49420001 – Pemphigus vulgaris
L10.0 – Pemphigus vulgaris
SNOMEDCT:
49420001 – Pemphigus vulgaris
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:01/09/2022
Last Updated:01/10/2022
Last Updated:01/10/2022
 Patient Information for Pemphigus vulgaris in Adult
Patient Information for Pemphigus vulgaris in Adult
Premium Feature
VisualDx Patient Handouts
Available in the Elite package
- Improve treatment compliance
- Reduce after-hours questions
- Increase patient engagement and satisfaction
- Written in clear, easy-to-understand language. No confusing jargon.
- Available in English and Spanish
- Print out or email directly to your patient
Upgrade Today

Pemphigus vulgaris in Adult
See also in: Anogenital,Oral Mucosal Lesion