PCT is the most common porphyria. Although the majority of cases are due to acquired or inherited uroporphyrinogen decarboxylase (UROD) deficiency, familial inheritance without a UROD mutation has also been described.
PCT is categorized into 3 types:
- Type 1 – Acquired, sporadic. Accounts for approximately 80% of cases. Factors causing type 1 PCT include iron overload, alcohol use disorder, smoking, hepatitis C virus (HCV) infection, use of certain drugs, and HIV infection.
- Type 2 – Autosomal dominant UROD mutation with incomplete penetrance. Accounts for approximately 20% of cases. Individuals with UROD mutations who do not have clinical symptoms are asymptomatic carriers.
- Type 3 – Familial mutation of genes other than UROD.
Susceptibility factors include use of alcohol and estrogen, smoking, chronic HCV infection, HIV infection (may be related to concomitant HCV infection), and UROD and hereditary hemochromatosis gene mutations. PCT has also been reported in conditions that cause oxidative liver damage (ie, diabetes mellitus and hepatic steatosis), pregnancy, advanced renal failure on dialysis (with associated iron overload), and myeloproliferative disorders. PCT has been reported in outbreaks with halogenated hydrocarbon exposure.
In both acquired and inherited PCT, reduction in UROD enzyme activity to 20% of normal is required before clinical manifestations occur.
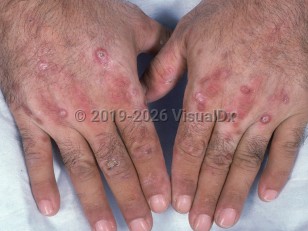
Once UROD activity is below 20%, porphyrins (mostly uroporphyrin) accumulate in the liver and the skin. Soret band (400-410 nm) of the visible spectrum activates porphyrins within cells and capillaries to enter an excited state. Photoactivated porphyrin subsequently releases protons to oxygen, and the resultant reactive oxygen species causes tissue damage. This manifests clinically as skin fragility, blistering, hypertrichosis, scarring, and sclerodermoid plaques. Photo-onycholysis may be seen. Patients may also notice urine discoloration.
There is an increased risk of hepatocellular carcinoma in patients with PCT, particularly those over 50 years of age; thus, routine monitoring is needed, particularly for those with other risk factors including cirrhosis, hepatitis C, or alcohol use disorder.