Erythropoietic protoporphyria (EPP) is a genetically determined deficiency of ferrochelatase, the enzyme catalyzing the final step of the heme biosynthetic pathway where heme is synthesized from iron and protoporphyrin IX.
X-linked protoporphyria (XLP) is caused by a gain-of-function mutation in the 5-aminolevulinate synthase (ALAS2) gene. ALAS2 catalyzes the formation of 5-aminolevulinate (ALA) from glycine and succinyl coenzyme A in erythrocytes.
In both, protoporphyrins build up in erythrocytes and in the bone marrow, skin, and liver. In the presence of visible light in the Soret band (400-410 nm) and oxygen, protoporphyrins release energy, and the resultant reactive oxygen species damage tissues. Erythrocyte protoporphyrin levels have been positively correlated with disease severity.
EPP and XLP are seen in all populations but are rare in Black people. Males and females are equally affected.
EPP and XLP present with similar clinical findings; XLP may have more severe manifestations. The hallmark is painful photosensitivity after brief exposures to sunlight. Symptoms are therefore worse in spring and summer and may begin in infancy or early childhood. Itch, tingling, or burning is experienced in sun-exposed skin after minutes to hours and progresses to pain with more prolonged exposure. Pain may last for hours up to a week and may be severe and incapacitating. Babies with EPP may cry on exposure to sunlight, and older children and adults may become sun avoidant, which can affect overall quality of life.
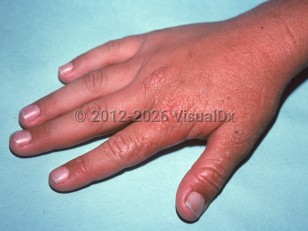
Edema and sometimes purpura of sun-exposed skin may be the only acute physical finding. Redness and petechiae may be seen. Vesicles and crusted erosions occur infrequently. Signs of chronicity include superficial scars on the nose and cheeks and radial scars around the mouth. The skin may appear thickened and wrinkled over the central face and small joints of the dorsal hands.
Hypochromic microcytic anemia may occur. Vitamin D deficiency may be secondary to sun avoidance. A minority of adult patients develop cholestatic hepatitis that may progress to liver failure.
Protoporphyria in Adult
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E80.0 – Hereditary erythropoietic porphyria
SNOMEDCT:
51022005 – Erythropoietic protoporphyria
E80.0 – Hereditary erythropoietic porphyria
SNOMEDCT:
51022005 – Erythropoietic protoporphyria
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:07/23/2023
Last Updated:07/24/2023
Last Updated:07/24/2023
Protoporphyria in Adult