Restrictive dermopathy is a rare lethal laminopathy inherited in an autosomal recessive fashion. A mutation in the zinc metalloproteinase (ZMPSTE24) gene or lamin A is responsible for the condition. Both sexes can be affected. Parental consanguinity increases risk.
Lamins are nuclear proteins that form a network of filaments beneath the nuclear membrane to maintain membrane integrity and control gene expression. Post-translation modification of prelamin A through proteolytic cleavage by zinc metalloproteinase enzyme (ZMPSTE24) results in the formation of mature lamin A. A defect in the enzyme (majority of cases) or in lamin A can lead to the clinical features of restrictive dermopathy.
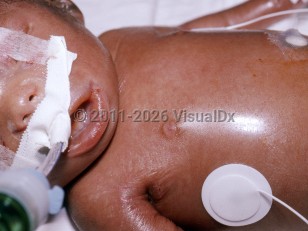
Decrease in intrauterine growth, restricted fetal movement, and polyhydramnios result in premature rupture of membranes and delivery of the child by 31 weeks of gestation. Typical clinical features at birth include tight, rigid, translucent skin; prominent superficial vessels; fixed, open mouth with retromicrognathism; small, pinched nose; multiple joint contractures; and erosions at flexure or pressure sites. Neonates with restrictive dermopathy die within few hours to a week after birth, usually because of respiratory insufficiency secondary to pulmonary hypoplasia.
Restrictive dermopathy
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q82.8 – Other specified congenital malformations of skin
SNOMEDCT:
400128006 – Lethal tight skin contracture syndrome
Q82.8 – Other specified congenital malformations of skin
SNOMEDCT:
400128006 – Lethal tight skin contracture syndrome
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/18/2022
Restrictive dermopathy