Retinoblastoma is the most common primary intraocular tumor in children. The incidence is approximately 1 in every 15 000 live births. Retinoblastoma may be inherited but is more commonly sporadic in occurrence. Approximately two-thirds of retinoblastoma cases affect only one eye and are not heritable. The tumor arises from a mutation of the retinoblastoma (Rb) tumor suppressor gene and may be inherited in an autosomal dominant pattern when there is a germline mutation. For retinoblastoma to develop, a second mutation must occur in retinal cells. Basic mechanisms were extensively reviewed in 2024. (For details, refer to Cobrinik; see References.) The tumor may be present at birth and is usually diagnosed by age 5. In the United States, the average age at presentation for a familial inherited retinoblastoma is 14 months, and 24 months for sporadic nongermline retinoblastoma. Retinoblastoma may be unilateral or bilateral, unifocal or multifocal.
The inherited form of retinoblastoma has no sex, race / ethnicity, or other predisposing factors, and it occurs equally in left and right eyes. The nonheritable form is more common in tropical and subtropical regions and indigent populations. Advanced paternal age has also been associated with the sporadic form of retinoblastoma.
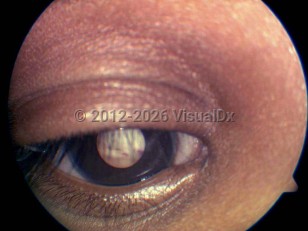
Leukocoria (white pupillary reflex) is by far the most common presenting sign in patients with retinoblastoma. In the United States, leukocoria as the presenting sign accounts for nearly 60% of all cases of retinoblastoma. Strabismus is the second most common presenting sign, seen in 20% of retinoblastoma patients in the United States. Patients with atypical signs and symptoms including a red, painful eye with glaucoma, cloudy cornea, poor vision, floaters, vitreous hemorrhage, and signs of orbital inflammation tend to be diagnosed with retinoblastoma at a later age, with more advanced disease.
In the developed world, over 95% of children will survive this cancer and over 90% will retain normal vision in at least one eye due to prompt diagnosis and improvements in treatment. Unfortunately, survival rates for less-developed nations lag at approximately 50%. Delay in diagnosis is the main factor affecting survival, with many of these patients presenting with orbital invasion or metastatic disease. Without treatment, children usually die within 2 years of diagnosis.
The main determinant for mortality is optic nerve invasion. Patients with invasion beyond the optic nerve or sclera or those with characteristic histologic features are at greatest risk for metastasis of retinoblastoma. Children with the inherited form of retinoblastoma have an increased risk of developing secondary cancers over their lifetime, including intracranial neuroblastic malignant neoplasm (trilateral retinoblastoma), osteogenic sarcoma of long bones, and sarcoma of soft tissues. Both the inherited and the sporadic form are associated with increased risk of cutaneous melanoma and nonmelanoma skin cancer. Although rare, cases of spontaneous regression of retinoblastoma have been reported.
Retinoblastoma - External and Internal Eye
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
C69.20 – Malignant neoplasm of unspecified retina
SNOMEDCT:
370967009 – Retinoblastoma
C69.20 – Malignant neoplasm of unspecified retina
SNOMEDCT:
370967009 – Retinoblastoma
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:05/06/2024
Retinoblastoma - External and Internal Eye