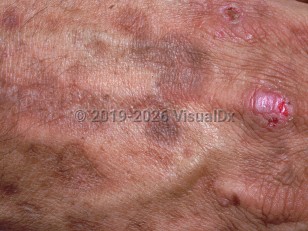
Porphyrias are a group of diseases resulting from defects / dysfunction in enzymes involved in heme biosynthesis. Several porphyrias have cutaneous features. Porphyrias with blistering cutaneous features include porphyria cutanea tarda (PCT) and hepatoerythropoietic porphyria. Porphyrias with nonblistering cutaneous features include erythropoietic protoporphyria and X-linked protoporphyria. Porphyrias that can have both blistering cutaneous features and acute neurovisceral attacks include hereditary coproporphyria and variegate porphyria (VP). Porphyrias with only neurovisceral symptoms without skin findings include acute intermittent porphyria and delta-aminolevulinic acid (ALA) dehydratase deficiency porphyria.
VP, also known as porphyria variegata, mixed porphyria, congenital cutaneous hepatic porphyria, and South African porphyria, is a blistering disorder caused by an autosomal dominantly inherited deficiency in protoporphyrinogen oxidase, a cytoplasmic enzyme involved in heme biosynthesis.
The prevalence is not known as many individuals are asymptomatic. Symptomatic VP has an estimated prevalence of 1:300 000 in Europe; this is higher in Southern Africans of Finnish and Dutch descent.
The spectrum of disease manifestation is broad, ranging from none to chronic photosensitivity and/or neurovisceral attacks. Acute neurovisceral attacks can be life-threatening. Homozygotes and compound heterozygotes tend to have more severe phenotypes.
The typical age of presentation is after puberty. Women are more commonly symptomatic for both skin and neurovisceral symptoms than men.
Cutaneous findings, which are the most common findings in VP, are caused by high plasma porphyrin levels and exposure of the skin to light with wavelengths in the range of 380-420 nm, with absorption peak at the Soret band, causing photoactivation, subsequent formation of reactive oxygen species, and tissue damage.
The exact pathogenesis of the acute attacks is poorly understood. It is thought that certain porphyrin precursors – such as ALA and porphobilinogen (PBG) – that are excreted by the liver in high amounts during attacks are potent neurotoxins.
Porphyrinogenic drugs (eg, barbiturates, phenytoin, other antiepileptics, rifampin), alcohol, smoking, hormonal changes, hormonal intake (eg progesterone), recurrent or chronic infections, gastric bypass surgery, and fasting or dieting (specifically carbohydrate restricting) may all precipitate an acute attack. Hepatic cytochrome p450 enzymes utilize heme as a cofactor; thus, medications degraded by the body through induction of these enzymes may accelerate heme synthesis and accumulation of the neurotoxic porphyrin precursors in these patients, thereby precipitating an acute attack. Such medications include barbiturates, estrogen / progesterone, griseofulvin, and sulfonamides, among others. The American Porphyria Foundation provides a Drug Safety Database Search that provides information about the interaction of specific drugs in patients with porphyria.
There is an increased risk of hepatocellular carcinoma in patients with VP, particularly those over 50 years of age; thus, routine monitoring is needed, particularly for those with other risk factors including cirrhosis, hepatitis C infection, or alcohol use disorder.
Emergency: requires immediate attention
Variegate porphyria
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E80.20 – Unspecified porphyria
SNOMEDCT:
58275005 – Variegate porphyria
E80.20 – Unspecified porphyria
SNOMEDCT:
58275005 – Variegate porphyria
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:02/06/2020
Last Updated:01/25/2022
Last Updated:01/25/2022
Emergency: requires immediate attention
Variegate porphyria