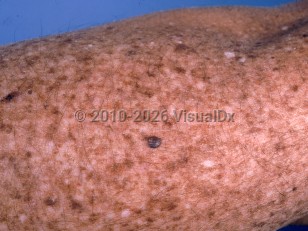
Individuals with XP usually have normal skin at birth but develop UV-induced changes including lentigines, blistering sunburns on sun exposure, dyspigmentation, and xerosis within the first several years of life. They later develop squamous cell carcinomas (SCCs), basal cell carcinomas (BCCs), melanomas, and ocular tumors (SCC and melanoma) at significantly increased rates. For patients with XP, the mean age of presentation with first nonmelanoma skin cancer (BCC or SCC) is 9 years, while the mean age of presentation of melanoma is 22 years. Additionally, patients with XP can also develop skin cancer on areas that are not typically exposed to UV light, most notably SCC of the anterior tongue. Approximately 90% of patients with XP will develop ocular manifestations. Ocular symptoms often present within the first decade of life. Progressive and irreversible neurologic degeneration is present in 20%-30% of patients with variable severity.
Patients with XP have a 10- to 20-fold increased incidence of internal malignancies such as brain, lung, hematopoietic, renal, and gastrointestinal tumors, which has been attributed to increased vulnerability to chemical and physical carcinogens, including UV light and tobacco smoke.
Patients may have mutations that result in defective global genome repair (GGR) or both GGR and transcription-coupled repair (TCR). When only GGR is defective, there is milder skin involvement and no neurologic abnormalities. There are 8 known complementation groups associated with XP, each with distinctive genetic loci and phenotypic variants:
- XPA – DDB1 protein, most common subtype in Japan and China
- XPB – ERCC3 gene, associated with Cockayne syndrome and trichothiodystrophy
- XPC – Endonuclease, most common subtype in the United States, Africa, and Europe, no neurologic sequelae
- XPD – ERCC2, associated with trichothiodystrophy and XP-Cockayne complex
- XPE – Only GGR defect, thus mild phenotype, no neurologic sequelae
- XPF – More common in Japan, only GGR defect, no neurologic sequelae
- XPG – Very rare, associated with Cockayne syndrome
- XPV – 30% of all cases, no neurologic sequelae