Agammaglobulinemia is a primary immunodeficiency in which B-cell development fails, leading to lack of circulating B-cells and hypogammaglobulinemia. This condition is most commonly inherited in an X-linked fashion and is known as X-linked agammaglobulinemia or Bruton agammaglobulinemia.
X-linked agammaglobulinemia is rare; the incidence is reported to be 1 per 250 000 live births in the United States. There is no known difference in frequency of this condition among various ethnic or racial groups, although diagnosis may be delayed in patients of color because skin lesions may be more difficult to discern in darker skim phototypes. Female carriers do not manifest clinical disease because of selective inactivation of the abnormal X chromosome in B-cells.
X-linked agammaglobulinemia results from a mutation in the gene for Bruton tyrosine kinase (BTK), found on the X chromosome. BTK is a nonreceptor cytoplasmic tyrosine kinase involved in B-cell receptor intracellular signaling, development, and differentiation. Loss-of-function mutations in BTK result in lack of mature B-cells and immunoglobulins of all classes. Fifty percent of the mutations are new, and these patients do not have a family history of the condition.
Less commonly, agammaglobulinemia is inherited in an autosomal recessive fashion and is referred to as autosomal recessive agammaglobulinemia. Autosomal recessive agammaglobulinemia is rare and constitutes at most 10%-15% of patients with agammaglobulinemia. Males and females are affected equally. In these cases, B-cell development fails because of mutations in multiple genes encoding mu heavy chain, immunoglobulin lambda-like-1 polypeptide, B-cell linker protein, and CD 79a.
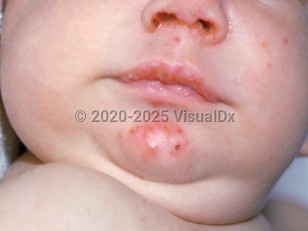
Patients with autosomal recessive agammaglobulinemia and X-linked agammaglobulinemia present similarly. Typically patients develop recurrent pyogenic infections between 6 months to 1 year of age, when maternal antibodies passed transplacentally disappear. These patients are susceptible to encapsulated organisms, most commonly Streptococcus pneumoniae, Haemophilus influenzae type B, and Staphylococcus. Sinopulmonary infections including otitis media, sinusitis, and pneumonia are most common, followed by skin infections, gastrointestinal infections, sepsis, meningitis, and osteomyelitis.
Patients with agammaglobulinemia handle fungal and viral infections normally except for enteroviruses; patients with agammaglobulinemia can develop meningoencephalitis and a dermatomyositis-like syndrome from systemic enterovirus infection. The most common enterovirus involved is echovirus followed by coxsackie virus and polio virus from live vaccine. Consequently, affected patients should avoid live polio vaccine. With regard to protozoa, patients with agammaglobulinemia are at increased risk for Giardia infection causing gastroenteritis.
In addition to infections, patients with agammaglobulinemia develop noninfectious skin and joint conditions. These patients can have an atopic dermatitis-like eczematous eruption that is not infectious. Rarely, pyoderma gangrenosum and noninfectious cutaneous granulomas have been reported in patients with agammaglobulinemia.
Twenty percent of patients develop a monoarticular nonseptic arthritis of large joints that may be confused with juvenile rheumatoid arthritis.
Patients with agammaglobulinemia are not at increased risk for autoimmune diseases, unlike patients with common variable immunodeficiency syndrome. The risk of lymphoreticular malignancies may be as high as 6%, and colorectal malignancies have been reported in patients with X-linked agammaglobulinemia. Failure to thrive is not commonly seen in affected patients aside from subsets that have growth hormone deficiency or develop bronchiectasis from recurrent pulmonary infections. When treatment with intravenous immunoglobulin (IVIG) is started before the age of 5, patients do better and have fewer infections. The exact life expectancy of these treated patients is not known but is thought to be at least into the sixth decade.
Congenital agammaglobulinemia in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
D80.0 – Hereditary hypogammaglobulinemia
SNOMEDCT:
116133005 – Congenital agammaglobulinemia
D80.0 – Hereditary hypogammaglobulinemia
SNOMEDCT:
116133005 – Congenital agammaglobulinemia
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Updated:01/12/2022