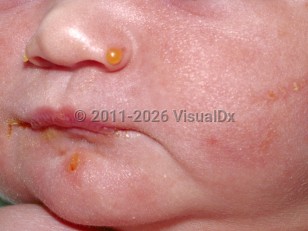
Inherited epidermolysis bullosa (EB) is a group of blistering disorders caused by defects in various components of the basement membrane zone (BMZ). Dystrophic EB (DEB) results from mutations in the COL7A1 gene, which encodes for type VII collagen. Type VII collagen is the chief anchoring fibril that forms an interconnected mesh adhering the lamina densa to the interstitial collagen fibrils in the sublamina densa. Mutations in COL7A1 result in the absence or reduced amounts of functional type VII collagen, leading to disruption of the BMZ between the lamina densa and the sublamina densa (papillary dermis). Clinically and histologically, this manifests in marked traumatic mucocutaneous fragility, causing a subepidermal (between the dermis and epidermis) split.
DEB is categorized based on the mode of inheritance: autosomal dominant DEB (DDEB) and autosomal recessive DEB (RDEB). There are currently 14 recognized subtypes of DEB. These variants are classified based on overall severity, clinical phenotype, ultrastructural and antigenic findings, and genotype. Depending on the subtype, DEB may be generalized at birth or during infancy with symptoms diminishing with age (seen with generalized DDEB). Other subtypes may eventually be clinically localized to specific anatomic sites (eg, acral, pretibial, nails) or have unusual distributions (eg, inverse, centripetal). EB pruriginosa is a very rare and typically intensely pruritic subtype. It can present shortly after birth or in infancy with mild blistering and erosions on acral surfaces, or in adulthood with excoriated, eroded, or lichenified papules and nodules and linear violaceous scars on the extensors. When generalized, DEB rarely may be life-threatening. In addition to blisters, erosions, and crusts, other common cutaneous findings include atrophic scarring, milia, fibrosis, joint contractures, and nail dystrophy. Scarring alopecia of the scalp may also occur.
Extracutaneous complications are common among patients with DEB, most notably those who have severe generalized RDEB (RDEB-sev gen, formerly known as Hallopeau-Siemens RDEB). The extent and severity of these complications vary among DEB subtypes. The most common sites involved include the external eye, oral cavity, esophagus, genitourinary tract, lower gastrointestinal tract, hands and feet, and bone marrow. Ocular manifestations include painful blistering, erosions, and surface scarring, which may lead to blindness. Oral findings include soft tissue scarring of the oral cavity and tongue, which may result in microstomia, and severe secondary caries formation, leading to premature loss of teeth. Gastrointestinal complications include stenosis, strictures, or webbing within the esophagus, chronic erosions within the small intestine (contributing to chronic malabsorption and malnutrition), constipation, gastroesophageal reflux disease (GERD), and painful anal fissures. Anemia, seen primarily in RDEB-sev gen, is multifactorial and usually severe. Other systemic comorbidities including malnutrition and osteoporosis can also occur. Marked growth retardation is seen in severely affected RDEB patients, as is delayed puberty. Progressive mutilating webbing of the fingers and toes (pseudosyndactyly) is common in RDEB, leading to deformities that may grossly impair use of the hands and limit ambulation. Patients with RDEB-sev gen are also at risk of renal failure and dilated cardiomyopathy, either of which may be fatal.
Blistering is worse in all EB patients during warm weather and may be exacerbated by concurrent systemic illnesses, including infection.
Death may occur in infants and young children with RDEB-sev gen, mainly as a result of sepsis. By age 30 years, over half of patients with RDEB, most notably those with RDEB-sev gen, develop cutaneous squamous cell carcinoma (SCC), which tends to occur in areas of chronic ulcerations or maximum scarring. Although these SCCs are typically histologically well differentiated, they may have an aggressive disease course. Although RDEB-associated SCCs may very rarely arise in childhood, most do not arise until after the middle or end of the second decade of life.
Related topics: EB simplex, generalized severe EB simplex, junctional EB, Kindler syndrome, EB acquisita
Dystrophic epidermolysis bullosa in Adult
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q81.2 – Epidermolysis bullosa dystrophica
SNOMEDCT:
254185007 – Dystrophic epidermolysis bullosa
Q81.2 – Epidermolysis bullosa dystrophica
SNOMEDCT:
254185007 – Dystrophic epidermolysis bullosa
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:03/04/2024
Last Updated:06/16/2025
Last Updated:06/16/2025
Dystrophic epidermolysis bullosa in Adult