While NF1 and NF2-SWN are monogenetic diseases and are inherited in an autosomal dominant pattern with up to 50% de novo mutations, SWN has a less well-defined inheritance pattern, with new mutations ranging in excess of 50%. As a group, these 3 neurocutaneous conditions make up part of the rare tumor suppressor syndromes, and all interfere with tumor suppressor function at the genetic level. They do not exhibit any sex or ethnic predilections. Because they are clinically and pathophysiologically distinct, they are considered separately.
NF1:
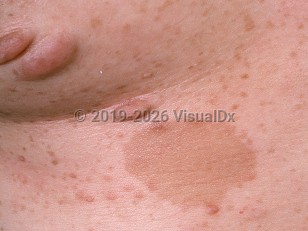
NF1, also known as von Recklinghausen disease, is a multisystem genetic disorder with hallmark cutaneous findings, including café au lait macules, neurofibromas, and axillary freckling. NF1 may affect the skin, nervous system, eyes, bone, and soft tissue. It is the most common autosomal dominant genetic disorder.
NF1 is often identified in childhood with the appearance of café au lait macules. The genetic defect is in a tumor suppressor gene on chromosome 17, which codes for neurofibromin, a RAS GTPase activating protein.
Diagnostic criteria for NF1:
The diagnosis of NF1 is made on clinical grounds based on 2 or more of the following features, if not better explained by an alternate diagnosis:
- Six or more café au lait macules greater than 5 mm in diameter in prepubertal patients and greater than 15 mm in diameter in postpubertal patients
- Two or more Lisch nodules (iris hamartomas)
- An osseous lesion such as sphenoid dysplasia or thinning of a long bone's cortex, with or without pseudoarthrosis
- At least 2 neurofibromas of any type or a single plexiform neurofibroma
- Freckling in the axillary or inguinal region
- Optic glioma (in early childhood)
- A first-degree relative with NF1
Other malignancies and tumors associated with NF1 include gliomas (especially optic pathway gliomas, occurring in 10%-15% of patients), pheochromocytomas, meningiomas, sarcomas, gastrointestinal tumors of neuroendocrine origin such as duodenal carcinoid tumors, and juvenile myelomonocytic leukemia. In addition to tumors and skin findings, patients may also have learning disabilities (30%-50%), skeletal anomalies, vasculopathies (including heart disease), and endocrinologic abnormalities.
Prompt ophthalmologic screening in patients with café au lait macules is recommended to diagnose NF1 and help prevent vision loss.
- Optic nerve gliomas are grade 1 pilocytic astrocytomas in NF1 that affect the optic nerve, chiasm, or optic tract. They are the most common intraorbital or cranial manifestation of neurofibromatosis, affecting 30%-40% of patients. Often asymptomatic, they are slow growing, locally invasive, and can progress to produce significant morbidity, although rarely mortality. Isolated asymptomatic optic nerve gliomas discovered during screening carry the best prognosis. Involvement of the chiasm, pulsatile proptosis, or extension along the visual pathway carries an unfavorable prognosis. Pulsatile proptosis, or the "Orphan Annie sign," is caused by absence of the sphenoid wing with a herniated encephalocele.
- Orbitofacial plexiform neurofibromas are in seen in 1%-4% of children with NF1. These are variants of benign peripheral nerve sheath tumors that form tangles of unencapsulated, highly vascular, and infiltrative neural tissue involving the eyelids, temporal region, and orbit and causing physical disfiguration, asymmetric refractive error, and mechanical ptosis with a high incidence of resultant amblyopia. They may degenerate into malignant peripheral nerve sheath tumors such as neurofibrosarcomas. Plexiform neurofibromas of the upper eyelid can be associated with ipsilateral glaucoma.
NF2-SWN is an inherited autosomal dominant genetic disorder that is approximately 10 times less common than NF1. Patients with NF2-SWN present primarily with vestibular schwannomas (schwannomas of the eighth cranial nerve), which are frequently bilateral. Hearing loss may be present. NF2-SWN is associated with a different suppressor gene mutation than NF1. It is located on chromosome 22 and codes for a protein called merlin. Patients have fewer café au lait macules than those with NF1, and they do not form Lisch nodules in the iris.
Diagnostic criteria for NF2-SWN:
- Bilateral vestibular schwannomas
- A first-degree relative with NF2-SWN in addition to either a unilateral vestibular schwannoma or any 2 of the following: meningioma, glioma, schwannoma, or juvenile posterior lenticular opacities
SWN is the least common of these 3 syndromes. SWN is more commonly sporadic in occurrence, but up to 25% are familially inherited in an autosomal dominant fashion. Individuals have nonintradermal schwannomas of the peripheral (90%) and spinal (75%) nerves and occasionally meningiomas (5%). While symptoms begin to develop in the teens or 20s, due to a typical diagnostic delay of approximately 10 years, most patients are not diagnosed until their third to fourth decades. Presenting symptoms are most often chronic pain (45%-50%), which is localized or diffuse and does not always correlate to the location of schwannomas, or an asymptomatic mass lesion (27%). Pigmentary changes and cutaneous schwannomas seen in NF1 and NF2-SWN are not characteristic of SWN, and patients with this syndrome do not exhibit learning disabilities. SWN may be diagnosed by purely clinical criteria or by combined clinical and molecular criteria.
Diagnostic criteria for SWN:
Clinical diagnosis –
- Two or more nonintradermal schwannomas, 1 pathologically confirmed schwannoma, and absence of bilateral vestibular schwannomas OR
- One pathologically confirmed schwannoma, unilateral vestibular schwannoma, or intracranial meningioma and 1 affected first-degree relative with SWN
- Two or more pathologically confirmed schwannomas or meningiomas and 2 or more tumors with 22q loss of heterozygosity and 2 different somatic NF2 mutations and none of the exclusion criteria OR
- One pathologically confirmed schwannoma or meningioma and a germline SMARCB1 or LZTR1 pathogenic variant
- A clinical diagnosis of NF2-SWN
- A germline NF2 pathogenic variant
- Schwannomas only within a radiation treatment field