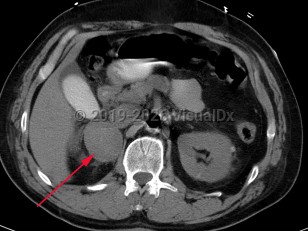
Pheochromocytoma (PCC) is a rare catecholamine-secreting neoplasm of adrenal origin; when it is extra-adrenal, it is referred to as paraganglioma. PCC is known as the "great masquerader" because of its similarity in clinical presentation to many other conditions. Up to 15% of patients may be asymptomatic. The most common presenting symptoms are hypertension (including labile hypertension), palpitations, increased sweating, severe headache, tremors, pallor, and dyspnea. Atypical symptoms include anxiety, abdominal pain, nausea, vomiting, constipation, insomnia, and weight loss. The prevalence of PCC in patients with hypertension is less than 0.2%. Rarely, paragangliomas may occur in the heart. Cardiovascular complications of catecholamine excess are rare but can lead to stress cardiomyopathy. Clinical presentation is characterized by transient duration of symptoms (eg, 15-20 minutes) that may recur several times a day. Physical exertion (moderate to extreme exercise) or bowel movements may provoke symptoms. Sometimes a pheochromocytoma is found incidentally during a workup.
The majority of PCCs are intra-adrenal and solitary. Approximately 10% of all catecholamine-secreting tumors will be malignant. PCC may occur at any age but has the highest prevalence in individuals aged 20-50 years.
Rarely, Pacak-Zhuang syndrome may be seen in patients with early-onset polycythemia and paragangliomas.
Treatment involves surgical removal of the tumor after adequate medical therapy is provided, generally with combined alpha- and beta-adrenergic blockade. Tumors can recur in less than 10% of patients. Surgery may help return norepinephrine and epinephrine levels to normal. Despite perioperative management, acute hypertensive crises may develop during surgical resection. Management of acute hypertensive crisis due to pheochromocytoma includes nitroprusside, phentolamine, or nicardipine.
In patients with malignant tumors who have undergone surgery, the 5-year survival rate is less than 50%.
Related topics: von Hippel-Lindau disease, multiple endocrine neoplasia type 2A, multiple endocrine neoplasia type 2B
Pheochromocytoma
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
C74.10 – Malignant neoplasm of medulla of unspecified adrenal gland
SNOMEDCT:
302835009 – Pheochromocytoma
C74.10 – Malignant neoplasm of medulla of unspecified adrenal gland
SNOMEDCT:
302835009 – Pheochromocytoma
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:03/06/2018
Last Updated:01/25/2023
Last Updated:01/25/2023
Pheochromocytoma