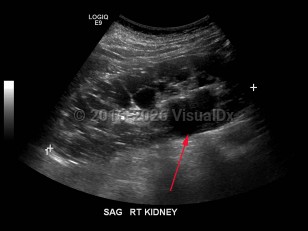
Polycystic kidney disease (PKD) is an autosomal dominant (or, less often, autosomal recessive) disorder characterized by numerous cysts in the kidneys, which may lead to end-stage renal failure. The autosomal dominant trait is associated with mutations in either PKD1 (which encodes polycystin-1) or PKD2 (which encodes polycystin-2). A minority of patients with PKD have a defect unrelated to PKD1 or PKD2. PKD1 mutations are more common and correlate with an earlier age of disease onset as well as more rapid decline in renal function.
Patients with autosomal dominant inheritance usually manifest symptoms at around 30 years of age; symptoms include abdominal pain, hematuria, and high blood pressure. Hypertension is very common and precedes the onset of overt renal dysfunction. Complications specific to renal cysts include cyst rupture, cyst infection, and nephrolithiasis.
Patients may develop brain aneurysms and liver cysts. Typically, this has a strong familial pattern, and the risk of cerebral aneurysm in patients with PKD is highest in those who have family members who have had aneurysms.
Children with autosomal recessive inheritance often manifest in the first decade of life and may develop progressive renal failure and hepatic fibrosis. The autosomal recessive trait is associated with mutations to the PKHD1 gene.
Treatment is supportive and focuses on blood pressure control, proteinuria management, and decreasing any other risks of kidney disease progression. No specific treatment has been shown to prevent or delay progression of renal dysfunction.
Polycystic kidney disease
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q61.3 – Polycystic kidney, unspecified
SNOMEDCT:
28770003 – Polycystic kidney disease, infantile type
765330003 – Autosomal dominant polycystic kidney disease
Q61.3 – Polycystic kidney, unspecified
SNOMEDCT:
28770003 – Polycystic kidney disease, infantile type
765330003 – Autosomal dominant polycystic kidney disease
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:02/20/2019
Last Updated:01/20/2022
Last Updated:01/20/2022
Polycystic kidney disease