Addison disease is the eponymous name given to primary adrenal insufficiency resulting from dysfunction of the adrenal glands. The estimated prevalence of Addison disease is approximately 5 per 100 000 with both sexes affected equally. Although patients of all ages can develop Addison disease, adults between the ages of 30 and 50 years commonly present.
Addison disease may be secondary to a wide variety of causes including autoimmune diseases, infections, vascular infarction, malignancies, and genetic / congenital etiologies. The most common cause of Addison disease in developed countries is autoimmune adrenalitis, which accounts for approximately 70%-90% of cases. Autoimmune adrenalitis causes the bilateral destruction of the adrenal cortex through both cell-mediated and humoral immune responses, often with identifiable antibodies against self-enzymes found in the adrenal cortex that are responsible for steroid production. Novel immune checkpoint inhibitors can cause checkpoint inhibitor-induced adrenalitis with a similar presentation.
Other causes of Addison disease include various infections, including HIV, tuberculosis, and fungal infections. Adrenal infarctions can be caused by bilateral adrenal vein thrombosis or hemorrhagic infarctions commonly associated with meningococcemia. Several malignancies can metastasize to the adrenals and disrupt adrenal function, including lung and breast cancers. Several drugs inhibit adrenal hormone synthesis including etomidate, azole antifungals, and suramin. Genetic causes of Addison disease that affect the adult population include autoimmune polyglandular syndrome, adrenoleukodystrophy, and congenital adrenal hyperplasia. See the child version of this summary for genetic causes that affect the pediatric population.
The destruction of the bilateral adrenal cortex leads to insufficient production of glucocorticoids, mineralocorticoids, and adrenal androgens. The manifestations associated with Addison disease are related to deficiencies of each hormone. They usually develop over time from the progressive destruction of the bilateral adrenals, but onset can occur suddenly from infarctions or trauma that cause acute adrenal destruction.
Early symptoms and signs of Addison disease include fatigue, weight loss, myalgias, arthralgias, salt cravings, postural hypotension, and nonspecific gastrointestinal symptoms (eg, nausea, vomiting, diarrhea, abdominal pain). In female patients, who depend more on adrenal androgens than male patients, there may be decreased axillary and pubic hair and loss of libido. Because these symptoms and signs are nonspecific, the diagnosis may go unrecognized until the patient presents with a life-threatening adrenal crisis.
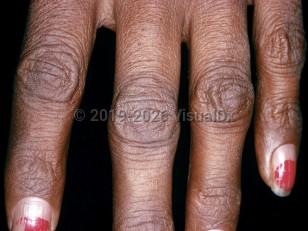
Over time, a significant fraction of patients with Addison disease will develop generalized skin and mucosal hyperpigmentation due to an increase in the production of pro-opiomelanocortin (POMC). POMC is a pro-peptide hormone that is cleaved to produce various products including adrenocorticotropic hormone (ACTH), beta-endorphins, and melanocyte-stimulating hormone (MSH). In the absence of adrenal hormone negative feedback to produce more ACTH, POMC secretion is elevated. The byproduct, MSH, stimulates melanin production, resulting in skin hyperpigmentation.
Patients with long-standing untreated Addison disease can have a wide range of psychiatric symptoms including depression, mild memory impairments, psychosis, and mania.
Addisonian Crisis:
Acute adrenal failure presents with severe abdominal pain accompanied by vomiting and diarrhea, low blood pressure, and loss of consciousness. This is a state of distributive shock and is potentially a life-threatening emergency that must be managed aggressively.
Addison disease in Adult
See also in: Oral Mucosal LesionAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
E27.1 – Primary adrenocortical insufficiency
SNOMEDCT:
363732003 – Addison disease
E27.1 – Primary adrenocortical insufficiency
SNOMEDCT:
363732003 – Addison disease
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
Drug Reaction Data
Subscription Required
References
Subscription Required
Last Reviewed:07/21/2020
Last Updated:03/03/2024
Last Updated:03/03/2024
Addison disease in Adult
See also in: Oral Mucosal Lesion