Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent or persistent candidal infections of the mucous membranes, skin, and nails that do not respond to conventional therapy. CMC arises due to defects in the host defenses against Candida albicans, the most common causative organism. Other Candida species, such as Candida parapsilosis, have also been implicated. Inadequate generation of interleukin-23 (IL-23), overproduction of IL-6, inefficient IL-17 activity, and an aberrant T helper 17 (Th17) pathway have been identified as mechanisms of host response dysfunction.
CMC may occur in familial forms with infection as the primary presentation or in association with various immune defects, where the infections are accompanied by additional features, such as endocrinopathy.
Familial forms of CMC have now been classified into 9 forms (although Type 5 has been reclassified as a distinct immunodeficiency syndrome).
Type 1: Autosomal dominant mutations on chromosome 2p predispose to dermatophytosis, alopecia, recurrent viral infections, tooth loss, thyroid disease, and mycotic aneurysms. The intravascular infections can cause symptoms mimicking a cerebrovascular accident. Some families have iron deficiency anemia and rosacea as well.
Type 2: Autosomal recessive mutations in CARD9 present with iron deficiency anemia, cutaneous hyperkeratosis, granulomatous reactions, and importantly, deeply invasive, potentially fatal candidal meningitis or encephalitis. See CARD9 deficiency.
Type 3: Infections with Candida predominate on the hands and feet with early onset and are associated with mutations on chromosome 11.
Type 4: In addition to infections with Candida, dermatophytes such as Trichophyton rubrum are often isolated. Onychomycosis and vulvovaginitis predominate in this autosomal recessive disorder attributed to mutations in CLEC7A.
Type 5: Reclassified as a primary immunodeficiency featuring infections with Staphylococci and recurrent bacterial respiratory infections.
Type 6: Autosomal dominant mutations in a single Argentinian family with CMC.
Type 7: Autosomal dominant mutations in STAT1 cause immunodeficiency type 31C with increased susceptibility to bacterial, viral, fungal, and mycoplasmal infections along with enteropathy, villous atrophy, delayed puberty, osteopenia, and autoimmunity. Intracranial aneurysms have been reported.
Type 8: Autosomal recessive mutations in TRAF31P2 present with mucosal candidiasis, folliculitis decalvans, staphylococcal infections, and atypical Candida species.
Type 9: Autosomal recessive mutations in IL17RC cause recurrent mucocutaneous candidiasis, impetigo, and onychomycosis without additional bacterial, viral, or other fungal infections.
Mutations in the autoimmune regulatory gene (AIRE) are specifically associated with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), which features recalcitrant candidal infections.
CMC may be a feature of immunodeficiency syndromes such as severe combined immunodeficiency, DiGeorge syndrome, hyper-IgE syndrome, CD25/IL2 receptor alpha mutations, or immune dysregulation polyendocrinopathy X-linked (IPEX). Nonimmune associations include keratitis-ichthyosis-deafness (KID) syndrome, acrodermatitis enteropathica, ectrodactyly-ectodermal dysplasia-clefting (EEC) syndrome, and multiple carboxylase deficiency.
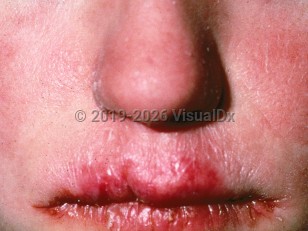
Features of CMC may develop in infancy. Variable involvement of the oral and genital mucosae, nails, and/or skin may be present in any sequence. Generally, infants present with either oral candidiasis (thrush) or diaper dermatitis. This is followed by onychomycosis and the possible later development of chronic disfiguring skin lesions, which appear as hyperkeratotic crusted areas on the face, scalp, and hands.
Specific syndromes may manifest additional clinical findings. APECED is associated with several disorders that include hypothyroidism, adrenal failure, gonadal failure, insulin-dependent diabetes mellitus, hypoparathyroidism, gastric parietal cell failure, keratopathy, vitiligo, and alopecia. Approximately half of these patients have 4 or 5 of the above manifestations. The most commonly associated endocrinopathy was hypoparathyroidism, followed by hypoadrenalism and ovarian failure.
Chronic mucocutaneous candidiasis in Infant/Neonate
See also in: Nail and Distal DigitAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
B37.2 – Candidiasis of skin and nail
SNOMEDCT:
234568006 – Chronic mucocutaneous candidiasis
B37.2 – Candidiasis of skin and nail
SNOMEDCT:
234568006 – Chronic mucocutaneous candidiasis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:01/25/2021
Last Updated:05/10/2023
Last Updated:05/10/2023
Chronic mucocutaneous candidiasis in Infant/Neonate
See also in: Nail and Distal Digit