This summary discusses dermatomyositis in children. Dermatomyositis in adults is addressed separately.
Childhood type dermatomyositis, also known as juvenile dermatomyositis (JDM), is an idiopathic multisystem inflammatory disorder where muscle inflammation is accompanied by a skin rash. It is typically first detected in children aged between 2 and 15 years.
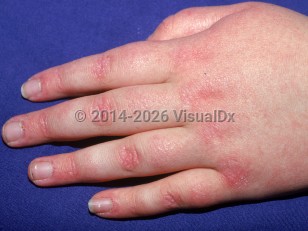
Cutaneous lesions may precede muscle disease by anywhere from weeks to more than 4 years and may be extremely pruritic. Exposure to sunlight may exacerbate the cutaneous lesions. Other characteristic features may include flat erythema of the upper back and posterior neck and shoulders (shawl sign) as well as a similarly presenting macular erythema of the anterior neck and upper chest (V sign) that can worsen with ultraviolet (UV) exposure. Patients may also have poikiloderma over the lateral hip (holster sign). Myositis (inflammation of muscle) usually develops insidiously, but sometimes abruptly, or may not occur at all (amyopathic or hypomyopathic dermatomyositis).
Muscle findings include symmetric proximal muscle weakness, usually of the shoulder and pelvic girdles.
In acute phases, pain and tenderness may occur and palpation may have a doughy feel. In addition, the patient's gait may be abnormal. Patients may complain of difficulty climbing stairs and brushing their hair. Upper palatal muscle and esophageal dysfunction may be noted, resulting in dysphonia, dysphagia, anorexia, and weight loss. Neck flexors are usually weak, and the patient may be unable to raise his or her head at all. Fever and fatigue are other common symptoms, and patients may also report arthralgia.
While children experience calcinosis cutis, muscle necrosis, and gingival and periungual telangiectasias more frequently than adults, JDM is a milder disease overall than adult dermatomyositis. There is a lower frequency of interstitial lung disease and lower overall mortality. Moreover, the association of malignancy with dermatomyositis is extremely uncommon in JDM.
Signs of severity in childhood include systemic vasculopathy, resulting in ulcerative disease of the skin and other organs (especially in the lungs or gastrointestinal tract), high severity of muscle disease, and endomysial fibrosis on muscle biopsy. The presence of these signs may indicate disease that is refractory to first-line treatment.
Dermatomyositis may be induced by medications, usually seen in adults, including hydroxyurea, penicillamine, interferon beta, statins, and ipilimumab. Acute onset / flares of dermatomyositis have been reported in association with ingestion of IsaLean, an herbal supplement.
Acquired lipodystrophy and acanthosis nigricans occurring in the setting of insulin resistance has infrequently been reported with JDM.
Juvenile dermatomyositis in Child
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
M33.00 – Juvenile dermatomyositis, organ involvement unspecified
SNOMEDCT:
1212005 – Juvenile dermatomyositis
M33.00 – Juvenile dermatomyositis, organ involvement unspecified
SNOMEDCT:
1212005 – Juvenile dermatomyositis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:06/25/2020
Last Updated:06/25/2020
Last Updated:06/25/2020
 Patient Information for Juvenile dermatomyositis in Child
Patient Information for Juvenile dermatomyositis in Child
Premium Feature
VisualDx Patient Handouts
Available in the Elite package
- Improve treatment compliance
- Reduce after-hours questions
- Increase patient engagement and satisfaction
- Written in clear, easy-to-understand language. No confusing jargon.
- Available in English and Spanish
- Print out or email directly to your patient
Upgrade Today

Juvenile dermatomyositis in Child