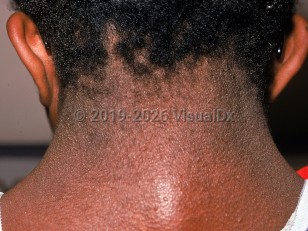
Noonan syndrome is a genetic disorder with a broad range of manifestations and disease severity. Patients classically present at birth or in early childhood with distinctive facial features, webbed neck, low occipital hairline, shield chest, short stature, coagulation defects, cardiovascular and lymphatic abnormalities, cryptorchidism in males, and generalized developmental delays.
Noonan syndrome has an estimated incidence of 1 in 1000-2500 live births in the United States. However, because Noonan syndrome is often misdiagnosed due to its highly variable presentation, the exact disease incidence / prevalence is currently unknown. Male patients are more commonly affected than female patients. Most cases of Noonan syndrome follow an autosomal dominant inheritance pattern.
Noonan syndrome belongs to a group of related disorders aptly named "RASopathies" based on perturbations in the RAS signaling pathway. Many genes have been found to be associated with Noonan syndrome, the most common being mutations in PTPN11, SOS1, RAF1, SHOC2, RIT1, and KRAS.
Some affected individuals only exhibit distinct facial features, while others carry the entire constellation of classic findings as outlined below. Due to the large overlap in clinical manifestations with Turner syndrome, Noonan syndrome is also known as pseudo-Turner syndrome.
The characteristic facial features of Noonan syndrome include ptosis, hypertelorism, downward-sloping palpebral fissures, epicanthal folds, ptosis, micrognathia, and low-set ears. Skeletal defects include shield chest, pectus deformities of the sternum, kyphosis / scoliosis of the spine, and generalized bone maturation defects resulting in short stature.
Approximately two-thirds of patients exhibit cardiac symptoms such as atrial septal defect, pulmonary stenosis, and hypertrophic cardiomyopathy. Many neonates with Noonan syndrome have lymphatic abnormalities resulting in lymphedema that manifests as inappropriate weight gain after birth. Blood clotting defects are commonly observed due to coagulation factor deficiencies, thrombocytopenia, and abnormal platelet functions.
Up to one-third of patients with Noonan syndrome have mild intellectual disabilities, but many have normal IQ. Male patients tend to experience abnormalities in the development of secondary sexual characteristics, and over 50% will be born with cryptorchidism. Female patients may experience delayed acquisition of secondary sexual characteristics but will usually develop normally.
Patients with Noonan syndrome have an elevated risk for malignancies in childhood including neuroepithelial tumors, neuroblastomas, acute myelogenous leukemia, and acute lymphoblastic leukemia. Other rare cancers, such as embryonal rhabdomyosarcoma, have been reported.
Noonan syndrome in Adult
See also in: External and Internal EyeAlerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q87.19 – Other congenital malformation syndromes predominantly associated with short stature
SNOMEDCT:
205824006 – Noonan's syndrome
Q87.19 – Other congenital malformation syndromes predominantly associated with short stature
SNOMEDCT:
205824006 – Noonan's syndrome
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:10/28/2019
Last Updated:04/11/2022
Last Updated:04/11/2022
Noonan syndrome in Adult
See also in: External and Internal Eye