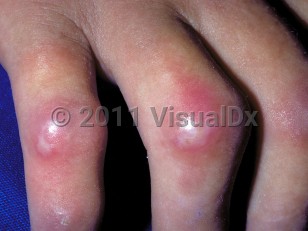
Scleroderma, or progressive systemic sclerosis, is an autoimmune connective tissue disease that involves sclerotic changes of the skin and may involve internal organs. While the etiology remains unknown, the disease is characterized by autoantibody production, collagen deposition, and vascular dysfunction. The disease is observed in all ages and is slightly more common in Black individuals and 3-4 times more common in women. The age of onset is usually between 30 and 50 years.
Scleroderma can affect the connective tissue of any organ, including the skin, gastrointestinal tract, lungs, kidneys, joints, muscles, heart, and blood vessels. Pulmonary disease is the leading cause of mortality. Additional common clinical features include esophageal dysfunction, primarily characterized by dysmotility, arthralgias, and Raynaud phenomenon. Less common manifestations include hypertensive renal crisis, pulmonary hypertension or interstitial lung disease, and cardiomyopathy.
There are 3 major clinical subsets of scleroderma:
- Limited cutaneous systemic sclerosis – distal skin sclerosis, Raynaud phenomenon, frequent severe late-stage complications such as pulmonary hypertension and gastrointestinal involvement. CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasias) refers to a subset of patients with limited scleroderma.
- Diffuse cutaneous systemic sclerosis – proximal extremity or trunk skin sclerosis, Raynaud phenomenon of shorter duration, high risk of renal crisis, and cardiac and lung fibrosis.
- Systemic sclerosis sine scleroderma – Raynaud phenomenon and systemic involvement without skin sclerosis.
Black individuals tend to have an earlier onset (aged 35-44 years compared with 45-55 years for all others), a more severe course, and increased mortality. This may be due to their likelihood of having diffuse, rather than limited, cutaneous systemic scleroderma. In addition, lung function is often worse. There is a greater prevalence of anti-topoisomerase I antibodies, anti-RNP, and anti-Ro antibodies in these patients, although the frequency of anticentromere antibodies is less compared with White individuals.
There are reports in the literature of drug-induced scleroderma. Most reports point to a sclerodermalike disease rather than true systemic scleroderma. See drug-induced sclerodermoid reactions for further details.

 Patient Information for
Patient Information for