The etiology and pathogenesis of DM is complex and involves genetic, environmental, and immunologic factors. Some evidence suggests that genetically susceptible individuals with certain human leukocyte antigen (HLA) types mount aberrant cellular and humoral responses after exposure to infection, malignancy, or drug ingestion.
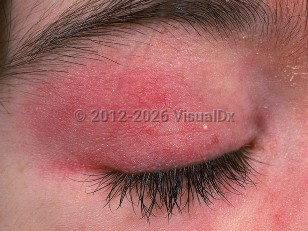
Clinical features of dermatomyositis can be categorized into cutaneous and systemic manifestations. Typical findings include a heliotrope rash, atrophic dermal papules of dermatomyositis (ADPDM, formerly called Gottron papules), shawl sign, holster sign, photosensitivity, flagellate erythema, poikiloderma, calcinosis cutis, and nail fold changes. Pruritus is also common.
Systemic manifestations of dermatomyositis include fatigue, malaise, myalgias, and the following:
- Musculoskeletal – Present in around 80% of patients with DM. There is proximal extensor muscle group inflammation that leads to acute or subacute weakness. Patients may have difficulty getting up from a sitting position or combing their hair. The myopathy is often painless.
- Gastrointestinal (GI) – Dysphagia due to pharyngeal and upper esophageal muscle involvement in up to 20% of DM patients.
- Respiratory – Diffuse interstitial fibrosis and acute respiratory distress syndrome are manifestations of pulmonary involvement. Some level of interstitial lung disease involvement will be present in 15%-30% of patients. Compromise of the accessory muscles of respiration also may contribute to poor ventilation and pneumonia. Dysphonia may reflect pharyngeal involvement.
- Cardiac – Most cardiac involvement in DM is subclinical and are arrhythmias and conduction defects including tachycardia, bradycardia, and bundle branch blocks. Congestive heart failure is the most commonly reported clinical cardiac manifestation of DM and is uncommon (~10%-15%).
- Malignancy – Adult patients with DM are at increased risk for internal malignancy. The reported frequency ranges from approximately 10%-50%, with the most accurate probable risk being 15%-25%. The period of highest risk is from 3 years prior to diagnosis to 3 years post diagnosis, possibly extending to 5 years following diagnosis. Ovarian cancer and colon cancer seem to predominate, with an additional increase in nasopharyngeal carcinoma in certain Southeast Asian groups. Other cancers commonly seen associated with DM include breast, lung, gastric, pancreatic, and lymphoma. Older age, male sex, dysphagia, and cutaneous ulceration have been reported to be associated with an increased cancer risk. The presence of anti-transcription intermediary factor 1 (anti-TIF1) immunoglobulin G2 (IgG2) is a biomarker for increased risk of malignancy in adults, with reported tumor rates of 20%-65%. The presence of antinuclear matrix protein 2 (anti-NXP2) antibodies in adults is associated with a tumor rate of around 35%.
- Autoimmune disease overlap – DM can occur in conjunction with systemic lupus erythematosus (SLE), mixed connective tissue disease, Sjögren syndrome, scleroderma, and rheumatoid arthritis. Scleroderma / CREST syndrome (calcinosis cutis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia) is the most common overlap syndrome, resulting in sclerodermatomyositis.
- Subtype: anti-synthetase syndrome (anti-SS) – Clinical triad of myositis, polyarthritis, and interstitial lung disease (ILD). Other characteristic findings include fever, Raynaud phenomenon, "mechanic's hands," and autoantibodies to aminoacyl-tRNA synthetases, predominately anti-Jo-1. This has a better prognosis than anti-MDA5-associated DM.
- Subtype: amyopathic DM (ADM) / clinically amyopathic dermatomyositis (CADM) – Approximately 20% of patients have no myositis, either clinically or by ancillary testing, for at least 6 months after presentation and are considered to have ADM. A smaller group, while having no clinical signs of myositis, will have evidence of subclinical muscle involvement on imaging studies and/or muscle testing such as electromyography (EMG) or biopsy. This group is deemed to have CADM. Many in this latter group develop clinically evident myositis later in the course of their disease.
- Subtype: Anti-melanoma differentiation-associated protein (anti-MDA5) antibodies are found in up to 65% of CADM cases and in 10%-20% of patients with classic DM. Their presence has been associated with the development of ILD, which may be rapidly progressive and portends a worse prognosis. Anti-MDA5 antibodies have also been associated with mucocutaneous ulceration, painful palmar papules, nonscarring alopecia, panniculitis, and arthritis.
- DM and malignant atrophic papulosis (Degos disease) are rarely associated.
- DM-associated environmental triggers include medications and supplements: hydroxyurea, tumor necrosis factor alpha (TNF-⍺) inhibitors, PD-1 inhibitors, CTLA-4 inhibitors, penicillamine, phenylbutazone, hydroxymethylglutaryl-CoA reductase inhibitors, lacosamide, cyclophosphamide, and omeprazole.

 Patient Information for
Patient Information for